
北京时间1月28日凌晨,《自然》(Nature)杂志发表了西北大学生命科学与医学部功能基因组学研究组严健教授团队及其合作者的最新研究成果《系统解析非编码DNA突变对转录因子结合的影响》,在国际上首次公布了该团队成功利用SNP-SELEX技术,分析人类基因组中近10万个常见的非编码位点突变对270个转录因子蛋白质结合的影响。
研究成果为揭示2型糖尿病等复杂疾病的遗传机制提供了重要的理论依据,是后基因组时代的一项突破性研究进展。SNP-SELEX技术为全面、快速解析代谢疾病、癌症等遗传疾病的分子机理,寻找诊断、治疗的靶标提供了新的思路与策略。

论文共同第一作者为西北大学严健教授、美国加州大学圣迭戈分校邱耘疆博士、巴西帕拉联邦大学的André M Ribeiro dos Santos博士。通讯作者为严健、任兵、Jussi Taipale教授。该成果由中国西北大学、美国路德维西癌症研究所和瑞典卡罗林斯卡医学院等单位共同合作完成。
什么是全基因组关联分析(GWAS)?
全基因组关联分析(GWAS)是广泛用于寻找复杂遗传疾病关联基因的重要手段。目前,全世界范围内已开展了4000多项GWAS研究,发现了超过10万个与各种疾病以及重要生理性状关联的基因位点。
但GWAS在找到基因序列差异与疾病风险关联的时候,并不能同时反映出这种关联背后的原理,这就阻碍了我们从关联中寻找预防和治疗这些疾病的分子靶标,成为一直困扰遗传学和医学研究者的一大难题。
近年来,越来越多的研究表明,这些疾病风险位点大部分位于基因组非编码区域,它们并不会改变蛋白质的序列与结构,而可能在基因表达调控上发挥作用。例如这些突变位点能够改变一些序列特异性结合蛋白与DNA结合的亲和力,从而影响下游目标基因的调控作用。但是,搞清楚一个位点突变的分子机理往往需要大量的时间和人力投入,单个突变一般需要有经验的研究组2-3年的时间完成,而且实验室之间、实验系统之间的差异还会干扰数据之间的可比性。考虑到目前已积累超过10万个疾病关联位点,而且GWAS还远远没有到达平台期出现饱和,随着研究手段的进一步改进,这样的位点还会越来越多。因此亟需开发一种可以高通量、系统性的办法来研究这些突变位点的分子功能。
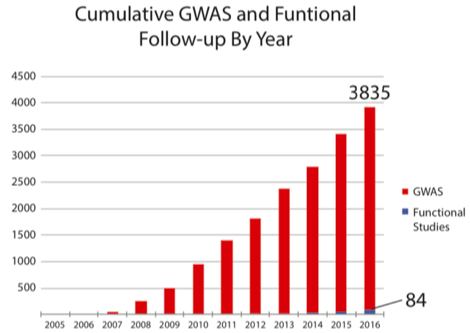
近年GWAS数据增长与功能研究增长对比
红色表示GWAS发现的位点随着时间推移的增加,蓝色表示对GWAS发现位点的功能研究的积累。
从HT-SELEX到SNP-SELEX技术8年的沉淀与改进
2013年,严健在JussiTaipale教授指导下发明了HT-SELEX技术,通过在体外表达纯化DNA结合蛋白,与人工合成的长度为40bp随机序列DNA片段结合,再经过多轮缓冲液的清洗去除非特异性结合,最后洗脱结合力强的DNA,经过高通量测序计数,定量分析转录因子的DNA序列结合特异性。
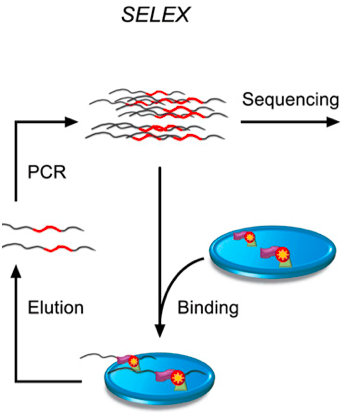
SELEX原理示意图
红色波浪线表示长度为40bp的双链DNA片段,每一轮循环表示一次富集过程,可以通过增加循环来提高富集度。
由HT-SELEX技术所产生的数据可以用于生成位置权重矩阵模型(PWM),该模型被广泛应用于预测转录因子能否与某DNA序列片段相结合。但经过对比发现,在预测SNP对于转录因子结合力影响时,PWM的效果并不理想,尤其是一些位于低亲和力结合位点的SNP。
为了摆脱对预测模型的依赖,在实验水平直接分析SNP对转录因子的影响,严健、任兵、Jussi Taipale教授于2015年再次联合提出改进HT-SELEX方法:在合成长度为40bp配体DNA时,不再使用随机序列的DNA,而以包含待研究的SNP及其附近的基因组序列取而代之,再利用HT-SELEX实验进行分析。研究团队利用该方法,定量分析了270个转录因子,在与95,886个非编码SNP结合时,不同等位基因序列(allele)与其结合强弱的差异,并记录了量化这些差异的大量数据。这些数据为解释疾病机理提供了重要的理论依据。
类似的研究手段可以进一步扩展到其他遗传疾病的研究中
以2型糖尿病风险位点SNP rs7118999为例,研究团队发现该位点可以影响转录因子HLF结合,并通过染色质长距离相互作用参与APOC3基因的表达调控,调节血脂水平。
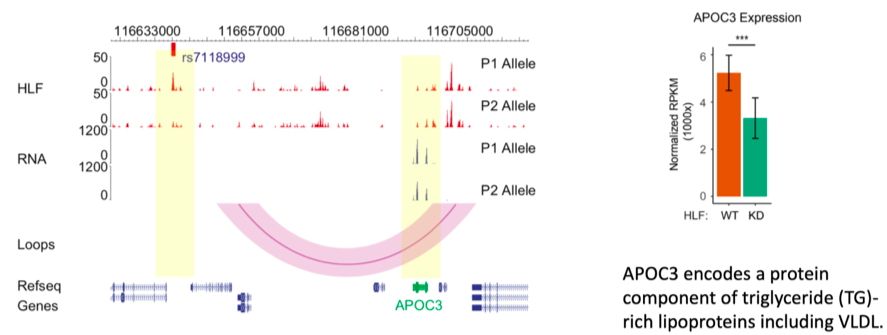
左图展示了在基因组浏览器中观察到的SNP rs7118999可以影响HLF转录因子的亲和力,从而通过三维基因组折叠的方式,引起下游APOC3基因表达的差异。右图展示了在肝脏细胞中敲低HLF会导致APOC3基因表达的下降,进一步验证了该团队的科学假设。
“之前的类似研究都是以单个或几个突变作为对象,这样要完全了解2型糖尿病这样同时受到几百上千个突变影响的复杂疾病,在短时间内是无法完成的,大大阻碍了开发治疗手段的进程。而这项研究一下子就解决了近10万个突变的分子机制问题,是一项重大突破。”严健说道。
该研究所产生的这些数据在后GWAS时代,对于揭示突变位点,尤其是非编码突变的分子机理,以及疾病的遗传模型有着重要的意义。
任兵表示:“目前我们的研究只包含了270个转录因子的SNP-SELEX数据,相比人类基因组中存在的1200-2000个转录因子还仅仅是一小部分。因此,我们的研究还会继续,期待能够全面了解非编码SNP的分子功能。”
严健介绍到:“以此为基础,我们相信类似的研究手段可以进一步扩展到其他遗传疾病的研究中,包括肠癌、前列腺癌等,将对解释这类疾病的遗传特性,找到临床诊断的分子标记物等工作都具有建议和指导作用。”

严健带领的功能基因组研究团队自2018年建立以来,在非编码基因组功能方向的研究取得一系列重大突破。此前,严健还以通讯作者身份在《自然方法》(Nature Methods)、《自然通讯》(Nature Communications)、《核酸研究》(Nucleic Acids Research)等国际知名期刊发表论文多篇。
为了方便其他研究人员使用这些珍贵的实验有关数据,文章中的所有SNP-SELEX结果都可以通过访问GVAT网页这一方式公开获得
(http://renlab.sdsc.edu/GVATdb/)。研究人员在该网页内输入感兴趣的转录因子名字或者SNP的rsID,便可快速找到与其相互作用的SNP或者转录因子,以及它们之间的量化影响。
原文链接:https://www.nature.com/articles/s41586-021-03211-0
该成果是西北大学生命科学与医学部自成立以来的首篇Nature杂志论文,也是以西北大学为第一作者单位在Nature和Science正刊发表的第16篇论文,是学校科学研究多点突破的标志性成果。
近年来,西北大学在持续推进“一院一策”改革的基础上,逐步打破学科边界,围绕学科交叉融合,促进学术协同创新,加快论证推进相关学科大部的建立。生命科学与医学部作为学校首家学部制试点单位,立足“大健康”学科集群,凝练和突出生物与医学学科优势特色,构建跨学科创新团队,建设大平台、承担大项目、产出大成果,逐步形成了面向国家需求、服务区域发展、彰显西大特色的“大健康”学科内涵式发展新路径。